An article written by Dr Edward Leatham, Consultant Cardiologist © 2025 E.Leatham
For busy people, or to tune in when on the move, Google Notebook AI audio podcast are available for this story beneath.
🧩 Why Do So Many People Have Raised LDL?
Raised LDL cholesterol, the principle (preventable) cause of coronary artery disease, is so common in modern societies that it’s almost considered normal — yet from an evolutionary and molecular standpoint, it’s anything but. Aside from the surprisingly small direct effect of high dietary intake of saturated fats, in most people there are three main causal systems that interact to determine an individual’s LDL set point — the level around which total LDL cholesterol naturally stabilises.
1️⃣ The PCSK9–LDL receptor pathway: the master regulator
The liver is the main consumer of circulating LDL cholesterol. It removes LDL particles from the bloodstream through specialised docking proteins on its surface called LDL receptors (LDLR).
Under normal conditions, LDLR captures LDL, internalises it, and recycles back to the liver surface to collect more LDL — maintaining low blood levels irrespective of dietary intake of fats.
Enter PCSK9 — proprotein convertase subtilisin/kexin type 9.
PCSK9 acts like a molecular off-switch for these receptors: when it binds to an LDLR, it marks it for destruction instead of recycling.
The more PCSK9 activity you have, the fewer LDL receptors survive — and the higher your blood LDL.¹
So, LDL level = balance between LDL production and LDL receptor recycling.
High PCSK9 activity raises the “set point,” because receptors are constantly destroyed and the liver must synthesise more cholesterol to meet its own needs.
2️⃣ Genetic and epigenetic variation: nature’s “set point mutations”
Some people are born with gain-of-function (GOF) mutations in the PCSK9 gene, making the protein overactive. These individuals recycle fewer LDL receptors, so their liver struggles to clear LDL from the blood. The result is lifelong hypercholesterolaemia and, often, early-onset coronary disease.²
Conversely, those with loss-of-function (LOF) variants have less PCSK9 activity and therefore more LDL receptors. Their LDL cholesterol can be 30–50 % lower for life — and their risk of coronary disease is dramatically reduced.³
Over 20 functionally validated GOF mutations and more than 40 LOF variants have been described.²,⁴,⁵ Their prevalence is low in absolute terms (each rare individually), but collectively, these genetic differences probably explain why LDL levels vary so widely between people eating the same diet.
However, genetics alone can’t explain why so many adults now have raised LDL. That’s where epigenetic and metabolic factors enter the story.
3️⃣ Metabolic inflammation, VAT, and the “secondary PCSK9 effect”
Modern research suggests visceral adipose tissue (VAT) — the fat stored around abdominal organs — can indirectly affect circulating LDL in several different ways:
i. Activation of the PCSK9 pathway.
VAT is not inert; it secretes cytokines such as IL-6, TNF-α, and resistin, which induce chronic low-grade inflammation in the liver and blood vessels.⁶
This inflammatory signalling triggers hepatic stress responses, including activation of transcription factors (SREBP-2 and HNF-1α) that increase PCSK9 expression.⁷
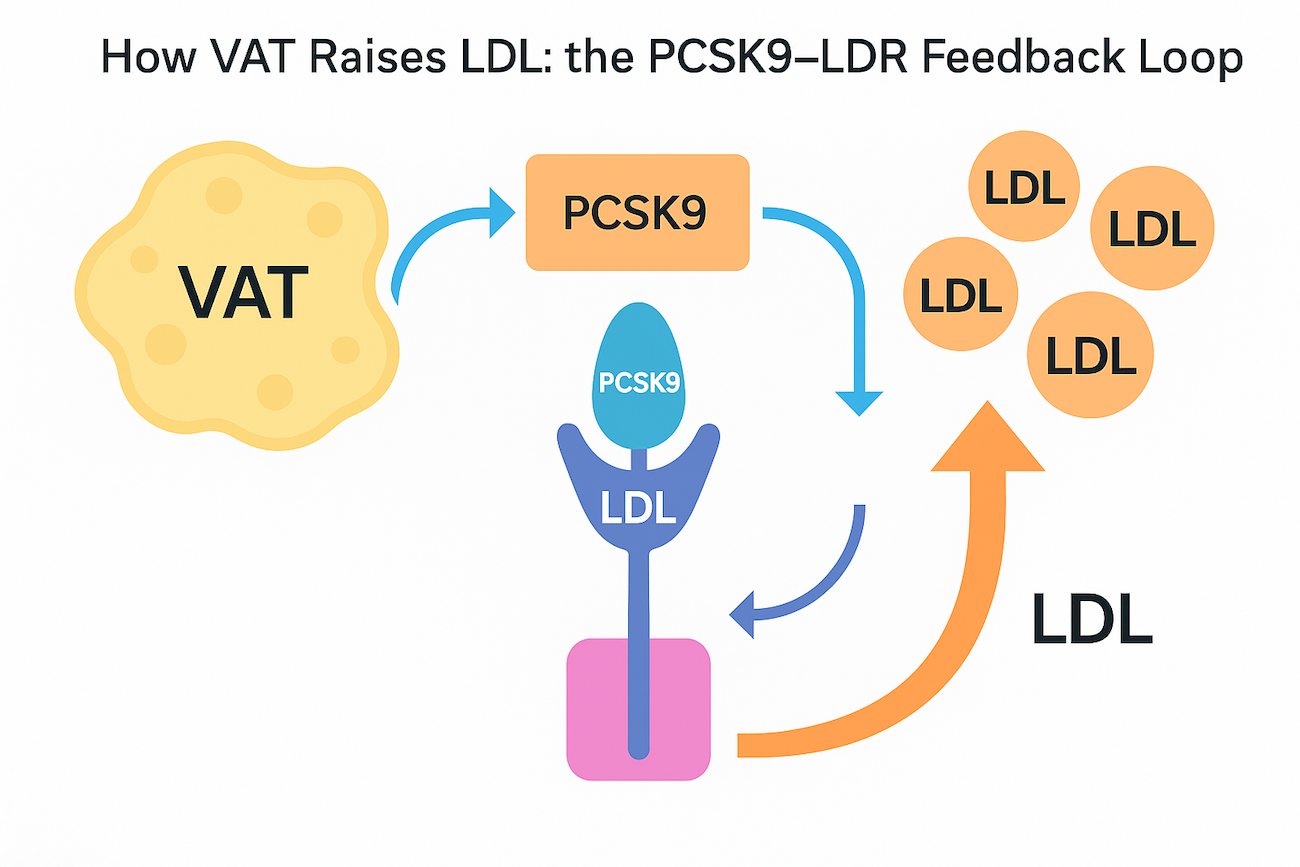
 Figure Caption: How VAT Raises LDL — the PCSK9–LDLR Feedback Loop
Figure Caption: How VAT Raises LDL — the PCSK9–LDLR Feedback Loop
Visceral fat releases inflammatory cytokines that activate liver stress pathways, increasing production of PCSK9, a protein that binds and destroys LDL receptors (LDLR). Once occupied by PCSK9, the LDL receptor is internalised and not recycled, reducing the liver’s ability to clear LDL from the blood. As receptor numbers fall, circulating LDL cholesterol rises, setting a higher “LDL set-point” even with a healthy diet. Reducing VAT through weight loss, GLP-1 therapy, or resistance training helps reverse this process, lowering both inflammation and LDL levels.
In other words, the body behaves as though it’s “under attack” and downregulates LDL receptor recycling to limit lipid flux and oxidative damage — an adaptive response that becomes maladaptive when sustained.
The result? An epigenetically raised LDL set point, even without a true genetic mutation.
ii/ VLDL and Triglyceride pathway (in insulin resistant states)
At super high visceral adipose tissue levels associated with high sugar /glycaemic index diets, the liver makes more triglyceride (TG) to help manage storage overload. TGs are transported in circulating VLDL, which is a molecule that circulates to drop off TGs in peripheral adipose tissue, where it shrinks in size to become small dense LDL (sd-LDL) These are the most atherosclerotic form of LDL – and represent roughly 20% of all LDL. They cannot be cleared as efficiency by LDLR so remain in the circulation for longer, where they can escape into the arterial wall at stressed points such as the coronary arteries. Once in coronary artery wall they get oxidised to cause coronary atherosclerosis.
Thus, people with raised VAT often have:
- higher PCSK9 levels,
- reduced LDL receptor activity,
- and persistently elevated LDL cholesterol despite modest dietary intake.
People with very raised VAT, especially those on high sugar diets with also often show subtle changes on simple blood tests :
- Low or falling HDL
- Rising or high blood Triglycerides (after an 8 hour fast)
- Rising or pre-diabetic HbA1c
This mechanism helps explain why obesity and metabolic syndrome so often coexist with raised LDL, and why lifestyle changes that reduce VAT — weight loss, resistance training, caloric restriction, low-glycaemic eating, or GLP-1 agonist therapy — lower risk and can normalise LDL, even without statins.⁸
The “metabolic vs genetic” interplay
In clinical practice, raised LDL usually arises from a one or more of:
- Genetic baseline (your inherited PCSK9–LDLR efficiency)
- Metabolic overlay VAT-driven inflammation, insulin resistance, and hepatic lipid overload driven by excessive intake of calories, especially carbohydrates that drive glucose and insulin which both increase PCSK9 and sdLDL via increased production of VLDL.
- High dietary intake of saturated and trans fats .
A person with a genetically normal PCSK9 pathway can therefore still develop high LDL (and sdLDL) if visceral fat, alcohol excess, or insulin resistance up regulate PCSK9 expression epigenetically.⁷
Conversely, someone with a loss-of-function PCSK9 variant may remain protected from LDL elevation even if overweight — though VAT may still drive inflammation and plaque activity through non-lipid pathways.
This duality — one pathway controlling how much LDL enters plaques, and another controlling how inflamed and unstable those plaques become — explains why modern prevention must target both LDL and VAT.
The new clinical synthesis
LDL lowering remains an evidence- based cornerstone of atherosclerotic prevention because it reduces the raw material for plaque formation.
However, visceral fat reduction not statin addresses insulin resistance and inflammatory activation that determine atherosclerosis and which possibly also contributes to inflammatory plaque rupture causing heart attacks. Lowering VAT also lowers LDL naturally by reducing PCSK9 activity.
Emerging evidence from GLP-1 receptor agonist (GLP-1RA) trials supports this.
The SELECT trial (2023) using weekly semaglutide in overweight non-diabetic adults showed a 20 % reduction in major cardiovascular events, despite only modest changes in LDL.⁹
The likely explanation is VAT loss — semaglutide reduces visceral fat by 25–30 %, dramatically improving insulin sensitivity and lowering inflammatory tone.¹⁰,¹¹
By doing so, it may indirectly modulate PCSK9, LDL receptor activity and sd-LDL while simultaneously calming coronary inflammation.
In this model:
- LDL drives atheroma formation (the supply chain of plaque cholesterol),
- VAT-driven inflammation and sd LDL overproduction determines plaque activity and rupture risk,
- and GLP-1 RAs, statins, and PCSK9 inhibitors tackle complementary ends of that process.
The clinical takeaway
For most people with raised LDL, the problem is not simply that they eat too much cholesterol.
It’s that their LDL receptor system is underperforming — sometimes genetically (GOF mutations in PCSK9 or APOB), but more often metabolically, due to VAT-driven hepatic inflammation and high insulin state that epigenetically raises PCSK9 expression and lifts production of ld LDL.
The modern liver, burdened with fat and inflammatory cytokines, acts like a warehouse manager who keeps re-ordering stock because the delivery system is faulty.
By addressing both the supply (LDL production) and the clearance (PCSK9–LDLR recycling), and by reducing VAT, we can restore the natural balance that evolution intended.
Does that mean there is no need for statins? If your future cardiovascular risk is low and you have a raised total LDL, then tackling high saturated fat intake and raised waist to height ratio (high VAT), is a logical move before contemplating statins. If you have established or early coronary heart disease, or a raised CaRi heartscore, where there is strong evidence of benefit from driving LDL to below 1.8 mmol/L, lowering VAT to lower PCSK9 and insulin-dependent pathway to lower ld LDL is an important adjunctive treatment alongside taking statins or other LDL lowering therapy. Based on available evidence to date, it is unlikely lowering VAT alone will achieve the target LDL required for the best long term outcome.
🩺 Clinical Application
For clinicians and patients interested in how these mechanisms translate into structured cardiometabolic care — including risk stratification, imaging decisions and escalation strategies — see my clinical overview:
Cardiometabolic Risk in 2026: From Self-Management to Structured Escalation
👉 https://www.scvc.co.uk/metabolic-health/cardiometabolic-risk-in-2026-from-self-management-to-structured-escalation/
📚 References
Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes. J Clin Invest. 2006;116(11):2995–3005. Available from: https://www.jci.org/articles/view/29383
Abifadel M, Guerin M, Benjannet S, et al. Identification and characterization of new gain-of-function mutations of PCSK9 in French families with autosomal dominant hypercholesterolaemia. Atherosclerosis. 2012;223(2):394–400. Available from: https://pubmed.ncbi.nlm.nih.gov/22683120/
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72. Available from: https://pubmed.ncbi.nlm.nih.gov/16554528/
Sarkar SK, Gonzalez M, Clark AJ, et al. Pathogenic gain-of-function mutations in the prodomain and C-terminal domain of PCSK9 inhibit LDL binding. Front Physiol. 2022;13:960272. Available from: https://www.frontiersin.org/articles/10.3389/fphys.2022.960272/full
Hoang Pham N, Truong P, Lao T, Le TA. Proprotein convertase subtilisin/kexin type 9 gene variants in familial hypercholesterolaemia: a systematic review and meta-analysis. Processes. 2021;9(2):283. Available from: https://www.mdpi.com/2227-9717/9/2/283
Bordicchia M, Spannella F, Ferretti G, et al. PCSK9 is expressed in human visceral adipose tissue and regulated by insulin and natriuretic peptides. Atherosclerosis. 2019;284:44–50. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6358804/
Dong B, Wu M, Li H, et al. Strong induction of PCSK9 gene expression through HNF1α and SREBP-2: mechanism for resistance to LDL-cholesterol lowering by statins in dyslipidaemic hamsters. J Lipid Res. 2010;51(6):1486–95. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3035512/
Després JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444(7121):881–7. Available from: https://www.nature.com/articles/nature05488
Lincoff AM, Ray KK, Kher U, et al. Semaglutide and cardiovascular outcomes in obesity without diabetes (SELECT). N Engl J Med. 2023;389(19):1735–46. Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa2307563
Wilding JPH, Batterham RL, Calanna S, et al. Impact of semaglutide on body composition in adults with overweight/obesity (STEP analyses). Diabetes Obes Metab. 2021;23(12):2588–99. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC8089287/
Nelson LW, Wadhwa V, Alemu BT, et al. Intrapatient changes in CT-based body composition after semaglutide treatment. AJR Am J Roentgenol. 2024;223(4):1–9. Available from: https://pubmed.ncbi.nlm.nih.gov/39230989/
Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. J Lipid Res. 2007;48(5):1084–91. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2711871/
Technical papers: located in Dr Leatham’s “VAT Trap” Digital Companion and Resources
- Why So Many PCSK9 Mutations Exist — Evolution, Immunity, and Trade-Offs
- Bradford Hill’s Criteria for Causation Applied to LDL Cholesterol and Coronary Heart Disease
- A Bradford Hill Appraisal of Raised Visceral Adipose Tissue and Coronary Heart Disease:
Related posts
- Cholesterol, LDL, and what we learnt from PCSK9 mutations in familial hypercholesterolaemia
- So what does determine your LDL (‘bad’) Cholesterol?
- LDL: the lower the better
- Visceral Fat, Mitochondria, and the Energy Trap: Why We Store Fat Where We Shouldn’t
- href=”https://www.scvc.co.uk/metabolic-health/visceral-fat-mitochondria-and-the-energy-trap-why-we-store-fat-where-we-shouldnt/”>Visceral Fat, Mitochondria, and the Energy Trap: Why We Store Fat Where We Shouldn’t
- Why everyone is talking about VAT