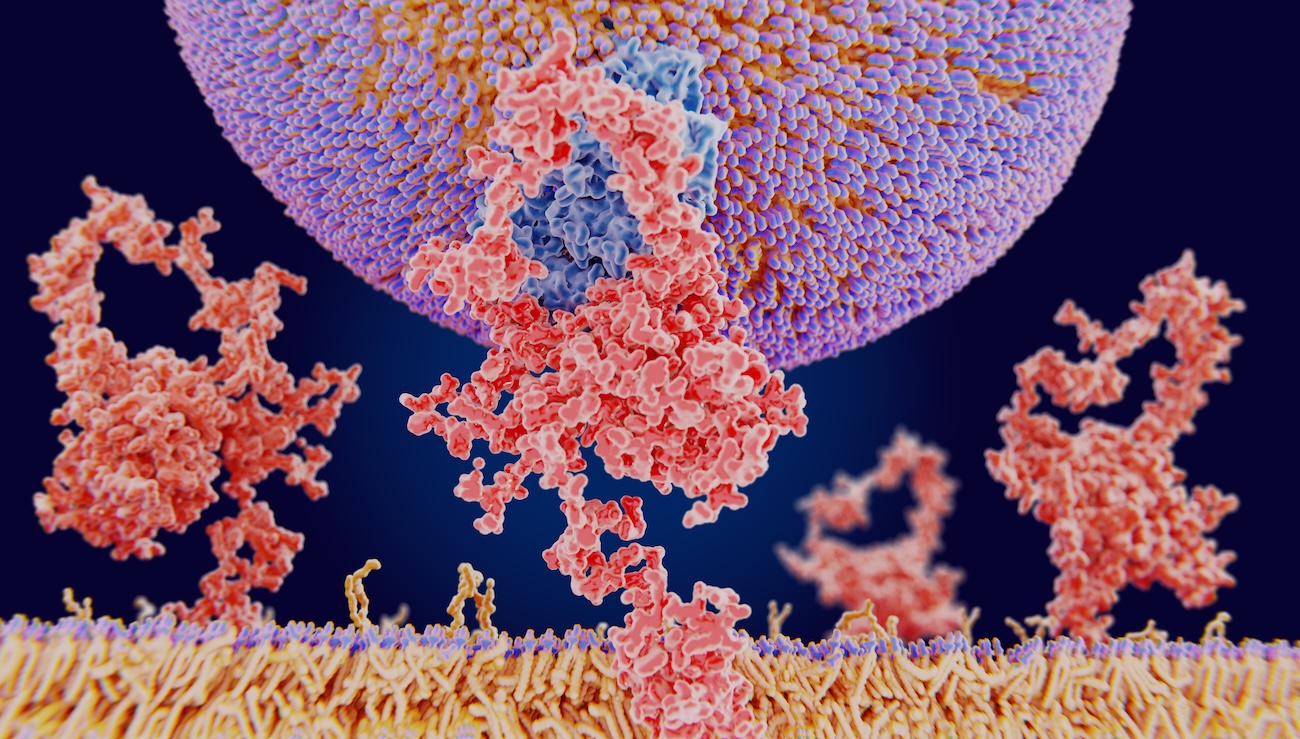
The 1st article in a series written by Dr Edward Leatham, Consultant Cardiologist for his VAT-TRAP series featured under this metabolic health blog.
For busy people, or to tune in when on the move, Google Notebook AI audio podcast and an explainer slide show are available for this story beneath.
Tags: VAT, Metabolic Health, LDL, PCSK9, VAT, VAT-TRAP search website using Tags to find related stories.
Many people have been told at some point that their cholesterol is “too high”. The usual response is to blame the food on our plates — butter, cheese, eggs, meat. Yet the story is far more fascinating than that, and much more forgiving.
Cholesterol is often portrayed as a villain, but it is essential for life. Every cell in your body contains cholesterol, which helps to repair membranes, produce hormones such as oestrogen, testosterone, and cortisol, make vitamin D, and synthesise bile — vital for the digestion and absorption of fats.¹
Because cholesterol is a fat-like substance, it cannot dissolve or travel freely in blood, which is mostly water. To move around, it must be packaged into microscopic transport particles called lipoproteins. One of these, LDL (low-density lipoprotein), acts as the main delivery vehicle, carrying cholesterol from the liver to cells that need it for repair or hormone production.
So, in small amounts, LDL cholesterol is completely normal and necessary. The problem only begins when too much LDL remains circulating in the blood for too long, particularly if it is small dense LDL (sdLDL) the small molecule variety of LDL, a byproduct of VLDL production, representing roughly 20% of total LDL.
The Liver: Control Centre of Cholesterol Traffic
The liver is the body’s main consumer and regulator of cholesterol. It uses cholesterol to produce bile and hormones, and it is also capable of making cholesterol itself.
Here is how the system works when everything is in balance:
- Cholesterol and fats from food are absorbed in the intestine, packaged into lipoproteins, and delivered to the liver.
- The liver’s surface is covered with LDL receptors — docking stations that recognise and internalise LDL particles to extract cholesterol for use or recycling.
- When dietary cholesterol is scarce, the liver simply synthesises its own to maintain supply.
- Some cholesterol is secreted into bile for digestion, but a proportion is reabsorbed in the gut and recycled.
This entire network is designed to ensure that cholesterol supply is steady. The body is remarkably efficient — it will not make what it can reuse.
Why So Many People Have High LDL
For many people, the system does not function perfectly either because the LDL receptors on liver cells are not always active or because the liver in response to high insulin high glucose state is programmed to accelerate energy storage by making and transporting triglycerides (TG), carried as VLDL which become low density LDL molecules.
In the first hepatic system an important regulatory protein called PCSK9 (proprotein convertase subtilisin/kexin type 9) is responsible for switching off LDLRs. PCSK9 binds to LDL receptors and directs them to be destroyed within the cell, rather than allowing them to recycle to the surface.² ³ When too much PCSK9 is present, fewer LDL receptors remain available to clear LDL from the blood. The result is simple: total LDL levels rise.
When that happens, the liver compensates for lack of cholesterol in needs by making even more cholesterol internally. This is why it is said that around 90% of the cholesterol in your blood is made by your liver, not absorbed from your diet.⁴
In families with inherited high cholesterol (familial hypercholesterolaemia, or FH), mutations in the PCSK9 gene have been identified which make this protein overactive. These are known as gain-of-function mutations — they increase PCSK9 activity and push LDL cholesterol to dangerous levels.⁵ For over 90% of people with raised LDL who do NOT have a mutation, there are alternative explanations outlined in a related article and for readers that are curious about the PCSK9 pathway take a look at my technical article linked to the VAT Trap book series on LDL Cholesterol.
In the second hepatic pathway, which is probably a great deal more important in populations with high prevalence of high sugar diets, obesity, insulin resistance and diabetes, the hepatic production of VLDL used to store surplus dietary intake (especially high glycaemic food and beverages), is likely to be the principle cause of a rise in small dense LDL. In this scenario, the principle pathogens are the high insulin state and much more atherogenic LDL particles, which are not cleared so efficiently by the liver LDLR.
An Evolutionary Mystery: Why PCSK9 Mutations Persist
If overactive PCSK9 raises cholesterol and promotes heart disease, why would evolution allow so many people to carry these mutations? It turns out there may be several reasons.
1. Protection Against Infection
One long-standing theory is that the LDL receptor — the same receptor that clears LDL cholesterol — can also act as an entry point for certain viruses and bacteria. By limiting LDL receptor availability, PCSK9 could have reduced vulnerability to infection.⁶
In an era before antibiotics, when infectious diseases killed huge numbers of children, genes that reduced infection risk would have conferred a significant survival advantage. A little more cholesterol later in life was a small price to pay.⁷
2. PCSK9’s Role in Immunity and Inflammation
Research has shown that PCSK9 does far more than regulate cholesterol. It also affects immune pathways, influencing inflammation and the way immune cells respond to infection.⁸
For example, PCSK9 can degrade immune-related receptors and alter how the body presents antigens to immune cells.⁹ ¹⁰ It may even modulate how macrophages (a type of white blood cell) handle lipids and inflammatory signals. This means that some mutations in PCSK9 could have made our ancestors better able to survive infection or injury.
3. A “Feast and Famine” Regulator
In ancient times, humans did not eat regular, balanced meals. There were periods of feast and famine. Some scientists believe that PCSK9 evolved as a metabolic brake, preventing the liver from being overwhelmed with cholesterol after periods of high-fat food intake. When food was scarce again, the body’s own cholesterol synthesis took over.¹¹
Today, with constant calorie availability, that same regulatory brake has become maladaptive — it keeps LDL receptors switched off even when we would prefer them to be active.
4. Neutral Drift and Genetic Tolerance
Another explanation is that many PCSK9 variants have only mild effects and did not cause problems until later in life — well after the age of reproduction. As a result, these variants were not removed by natural selection.
The PCSK9 protein also has a complex structure that allows many different small mutations to subtly alter its behaviour without destroying it. Over thousands of generations, this created a wide variety of PCSK9 activity levels within the population.
Why Low-Fat Diets Rarely Fix High LDL
Given this biology, it becomes clear why reducing dietary fat alone rarely normalises cholesterol levels. When you lower cholesterol intake, the liver simply increases its own production. In most people, a strict low-fat diet can lower LDL cholesterol by no more than about 10%. Recently, a top lipidologist told me on average that 10% of people get only a 4% reduction, although in my experience there is wide variation, depending on factors such as dietary intake of saturated fats (especially important in some people on keto or Atkins diet), insulin resistance and visceral adipose tissue. In fact, the only way to really find out is for each person to undertake their own ‘N of 1 trial’ and remeasure their lipids 3 months after starting their new diet.
The other problem that can present is that cutting fat often leads people to replace calories with carbohydrates. For those with a carbohydrate-sensitive phenotype (CSP), this can raise insulin levels, stimulate liver fat production, and increase visceral fat leading to more VLDL, sLDL and PCSK9 production— undoing much of the intended benefit.
However, there is considerable body of evidence that, in order to prevent cholesterol building up in the arterial wall, we generally need circulating total LDL below 1.6 mmol/L (62 mg/dl), and in high risk cases, ideally even strive below 1.0 mmol/L (38 mg/dl) for those at high cardiovascular risk.¹,²
Diet alone very rarely achieves this.
Modern Therapies That Work With — Not Against — Your Biology
Modern treatments target the cholesterol system at different points, each designed to restore the liver’s ability to remove LDL from the blood.
1. Statins
Statins block the enzyme HMG-CoA reductase, a key step in cholesterol production in the liver. When liver cholesterol levels fall, the liver reacts by increasing LDL receptor production, allowing it to pull more LDL out of circulation.⁸
This indirect mechanism “tricks” the liver into cleaning the blood, but because it affects cholesterol synthesis in all cells, some people experience muscle aches or mild effects on glucose control.
2. Ezetimibe
Ezetimibe prevents cholesterol from being reabsorbed in the gut, forcing the liver to draw more from the blood to meet its needs.¹,⁴ It is often used alongside statins for an additive effect.
3. PCSK9 Inhibitors
These injectable drugs directly block the PCSK9 protein, preventing it from degrading LDL receptors. This allows the receptors to remain active and continuously clear LDL from the bloodstream. Clinical trials show that PCSK9 inhibitors can reduce LDL by 50–60%,¹,⁵ with parallel reductions in heart attack and stroke risk.¹⁶
Newer therapies such as siRNA-based drugs (for example, inclisiran) silence PCSK9 gene expression in the liver, providing the same effect with only a few injections per year.
4. VAT-lowering programs and GLP-1 mimetics
In recent years, trials of GLP-1 mimetic drugs have established major survival benefits in non diabetics with coronary heart disease. The exact mechanism of benefit remains a matter for debate and intense scientific scruity, however it seems likely that the principle effect of these drugs is their impact on reducing visceral adipose tissue, reducing insulin resistance and sdLDL. This finding is the reason why cardiologists are increasingly including waist to height ratio and low dose VAT CT to risk-profile our higher risk patients alongside lab bloods, and also why we offer VAT reduction programmes with or without GLP-1 mimetics to treat metabolic cause of heart disease alongside traditional modifiable risk factors such as hypertension and raised LDL.
The Takeaway: A Story of Survival and Mismatch
The liver’s PCSK9 system is a brilliant evolutionary design — a fine-tuned mechanism that protected our ancestors against infection and nutritional extremes. But in modern life, where food is plentiful and people live into old age, our liver’s storage and cholesterol pathways can become a liability.
High LDL cholesterol can be the result of high sugar and carbohydrate diet associated with high VAT and is not simply the result of high dietary intake of saturated fats. However in patients with healthy metabolism without insulin resistance it may more often be the legacy of an ancient survival mechanism now out of sync with modern longevity. The good news is that with the right understanding — and, when necessary, targeted therapies — we can rebalance the system and protect our arteries for life.
References
- Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. J Lipid Res. 2007;48(5):1084–91.
- Seidah NG, Awan Z, Chrétien M, Mbikay M. Multifaceted biology of PCSK9. Endocr Rev. 2022;43(3):558–85.
- Rosenson RS, Shapiro MD, Yeang C, Fazio S. The evolving future of PCSK9 inhibitors. J Am Coll Cardiol. 2018;72(13):1477–89.
- Abifadel M et al. Identification and characterisation of new gain-of-function mutations of PCSK9. Atherosclerosis. 2012;223(2):394–400.
- Di Taranto MD et al. Identification and in vitro characterisation of two new PCSK9 gain-of-function variants. Sci Rep. 2017;7:15282.
- Labonté P et al. Low-density lipoprotein receptor as a hepatitis C virus entry cofactor. J Virol. 2009;83(23):12671–82.
- Mbikay M, Mayne J, Seidah NG. The biological relevance of PCSK9: when less is better. Biochem Cell Biol. 2022;100(6):447–63.
- Bao X et al. Targeting PCSK9: novel molecular and therapeutic insights. Signal Transduct Target Ther. 2024;9:205.
- Ricci C et al. PCSK9 and innate immunity: interactions with toll-like receptors during infection. Front Immunol. 2023;14:1175421.
- Demers A et al. PCSK9 induces CD36 degradation and affects macrophage lipid uptake. J Biol Chem. 2015;290(6):3446–57.
- Seidah NG et al. Multifaceted biology of PCSK9. Endocr Rev. 2022;43(3):558–85.
- European Society of Cardiology. 2023 Guidelines for Cardiovascular Disease Prevention. Eur Heart J. 2023;44(36):3726–3828.
- Ballantyne CM et al. Ezetimibe: a clinical overview. Clin Lipidol. 2017;12(3):615–28.
- Lagace TA et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes. Proc Natl Acad Sci USA. 2006;103(15):5489–94.
- Ridker PM et al. Evolocumab and clinical outcomes in patients with cardiovascular disease (FOURIER trial). N Engl J Med. 2017;376(16):1527–39.
Small Dense LDL: Scientific Background, Clinical Relevance, and Recent Evidence Still a Risk Even with ‘Normal’ LDL-C Levels
Technical papers: located in Dr Leatham’s “VAT Trap” Digital Companion and Resources (free to download)
- Why So Many PCSK9 Mutations Exist — Evolution, Immunity, and Trade-Offs
- Bradford Hill’s Criteria for Causation Applied to LDL Cholesterol and Coronary Heart Disease
- A Bradford Hill Appraisal of Raised Visceral Adipose Tissue and Coronary Heart Disease:
Related posts
- Cholesterol, LDL, and what we learnt from PCSK9 mutations in familial hypercholesterolaemia
- So what does determine your LDL (‘bad’) Cholesterol?
- LDL: the lower the better